

Mancata penetranza? Mosaicismo?
Un altro dato che ancora non è possibile chiarire è l’esistenza di malati che non presentano, o nei quali comunque non è stata evidenziata, una mutazione del gene GNAS.
A complicare ulteriormente le cose, le mutazioni GNAS, le stesse, possono portare anche ad altre patologie come l’AHO e il POC, anche nella stessa famiglia!
Quest’ultimo dato si spiega con l’imprinting (espressione differenziata di un gene in rapporto alla sua origine paterna o materna), e cioè in questo caso con l’assenza di espressione del gene GNAS sottoposto ad imprinting quando l’allele presente sul cromosoma omologo subisce la mutazione dello GNAS. Per cui a seconda se la mutazione è portata dall’allele paterno o materno si avrà una differente malattia, e questo può essere vero solo se di regola il gene è espresso dall’allele materno, almeno nella maggior parte dei tessuti, dato questo che spiegherebbe perché la trasmissione materna della mutazione causa le manifestazioni più gravi come la sindrome di Albright da PHP-Ia e la trasmissione paterna lo PseudoPseudoIpoparairoidismo, oltre a POH e POC.
In realtà la cosa è ancora più complessa perché c’è una espressione predominante del Gsα materno solo in certi tessuti. Si deduce, da quanto andiamo dicendo, che l’espressione e l’imprinting di GNAS è abbastanza complesso e probabilmente non del tutto chiarito.
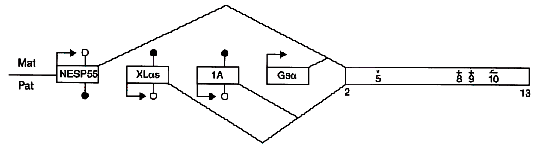
| Fig. 3: Organizzazione gene GNAS con i suoi 4 promoters : NESP55 e Gsα sono espressi dall’allele materno, XLas e 1A sono espressi dall’allele paterno! |
GNAS ha 4 primi esoni alternativi che corrispondono a 4 differenti promoters
che si congiungono ad un gruppo comune di esoni (2 – 13). Gsα è codificato dall’esone 1 e biallelicamente espresso nella maggior parte dei tessuti, NESP55 che è sintetizzato dall’allele ereditato per via materna, l’esone XLαs e 1A trascritti dall’allele ereditato per via paterna.
Resta comunque certo, nonostante questa complessità evidentemente ancora non risolta, che le mutazioni somatiche inattivanti di GNAS quando ereditate per via materna danno PHPIa, quando ereditate per via paterna danno PPHP o POH o POC. Lo studio della famiglia di cui abbiamo detto prima è l’esempio più lampante di quanto abbiamo or ora descritto: la prima generazione da padre portatore di mutazione GNAS presenta 5 figlie femmine affette da POH; la seconda generazione, da femmine affette da POH, presenta 4 figli maschi affetti da PHPIa!
Recentemente uno studio del POH Collaborative Research Group ha messo in evidenza come nei casi sporadici di POH la mutazione dello GNAS è sempre insorta sull’allele paterno.
Altre considerazioni vanno fatte nel cercare di dipanare questo enigma, prima di tutto la proteina Gsα viene normalmente espressa biallelicamente in molti tessuti (1 solo allele cioè è insufficiente a produrla: Aploinsufficienza), ma non in tutti! Questo spiegherebbe in parte la variabilità fenotipica della patologia da GNAS.
È vero che l’imprinting genomico fa si che prevalga la comparsa dell’una o dell’altra patologia, ma sono descritti casi con patologie associate!
Probabilmente nell’estrinsecazione fenotipica intervengono altri prodotti dello stesso gene, trascritti dai promoters di cui abbiamo già detto.
Ma è anche probabile che intervenga un secondo gene o anche più di uno, ed è possibile anche l’effetto di altri loci adiacenti.
Tutto questo ci porta a pensare la POH come solo un estremo di un ampio spettro di condizioni patologiche con ossificazioni, geneticamente correlate.
La diagnosi
La POH è identificata con il n. 166350 nella classificazione/data base OMIM, la diagnosi al momento è preminentemente clinica e, riassumendo quanto abbiamo rilevato nelle pagine precedenti, possiamo definire così i criteri diagnostici che ci permettono di fare diagnosi di POH:
- il peculiare quadro dermatologico iniziale con elementi nodulari di durezza lapidea,
- la progressione dell’ossificazione dai piani superficiali verso i tessuti profondi,
- il caratteristico aspetto radiologico figurato, a rete, a merletto, a bozzolo, con l’assenza di alterazioni scheletriche primitive,
- la normalità degli esami ematologici ed endocrinologici con assenza di squilibri ormonali, salvo l’aumentata attività della fosfatasi alcalina nelle fasi di più intensa produzione di ossificazioni,
- la normalità dell’aspetto morfologico e della intelligenza,
- il tipico quadro istologico con ossificazione membranosa diretta,
- la presenza di una mutazione inattivante del gene GNAS (probabile!),
- la familiarità per patologie con ossificazione (AHO, POH, POC)(possibile!),
La diagnostica differenziale
Nella diagnostica differenziale di questa patologia vanno considerate le altre patologie con ossificazione eterotopica.
Facile è escludere tute le forme secondarie a traumi, malattie vascolari, artropatie.
Tra le forme primitive dobbiamo tener presente la Fibrodisplasia Ossificante Progressiva (MIM 135100) e la Osteodistrofia di Albright che può corrispondere allo PHPIa (MIM 103580) o allo PPHP (MIM 300800). Ovviamente va tenuto presente l’Osteoma cutis o POC (MIM 166350, cioè lo stesso identificativo che per la POH!).
La FOP è una malattia geneticamente determinata caratterizzata da trasmissione autosomica dominante, in questo caso il gene mutato è l’ACVR1, individuato non più tardi della fine di aprile del 2006.
In questa patologia c’è una osteogenesi eterotopica progressiva a partenza in genere dai cingoli e dalla nuca, che inizia con una flogosi muscolare a cui fa seguito la comparsa di noduli dolorosi di tessuto fibroproliferativo coinvolgente tendini, legamenti e il tessuto connettivo dei muscoli scheletrici.
Successivamente c’è ossificazione di questi tessuti, una ossificazione che dal punto di vista istologico è di tipo encondrale con fase cartilaginea e presenza di isole cartilaginee nei preparati istologici.
L’inizio della patologia è nella prima decade di vita con immobilizzazione progressiva delle articolazioni dello scheletro assiale e degli arti con caratteristici pattern di progressione: dorso à ventrale; assiale à appendicolare; cranio à caudale e prossimale à distale.
Nei soggetti affetti è presente una caratteristica malformazione bilaterale congenita degli alluci. L’ossificazione può essere innescata da un trauma di punta, come un ago ed infatti talvolta ricorre nella storia clinica di questi soggetti la comparsa di prime manifestazioni a seguito di iniezioni, anche vacciniche; comunque può iniziare anche spontaneamente.
Da questo processo ossificativo progressivo sono risparmiati i muscoli estrinseci dell’occhio, il diaframma, il cuore e i muscoli lisci; anche la cute è sempre risparmiata.
L’escissione chirurgica delle ossificazioni ectopiche è non solo inutile, dato il carattere congenito e progressivo della patologia, ma anche dannoso in quanto il trauma stesso operatorio porta a stimolare nuova produzione di osso ectopico.
L’evoluzione è praticamente sempre fatale con episodi di riaccensioni acute improvvise e importanti (flare-up). La morte prematura di questi soggetti spesso è il risultato di insufficienza respiratoria, da restrizione della gabbia toracica, o di inanizione dovuta alla grave anchilosi della mandibola che spesso si instaura e che non permette in maniera assoluta l’alimentazione.
Nella sindrome di Albright dovuta a PPHP ritroviamo oltre alla formazione di osso ectopico che interessa soltanto cute e sottocute, anche una classica morfologia somatica e clinica generale caratterizzata da obesità, faccia a luna piena, bassa statura con alterato rapporto peso/altezza, brachidattilia per la presenza di ossa metacarpali e metatarsali corte, ritardo mentale; oltre a ciò possono essere presenti erosioni periostee caratteristiche. Anche qui però possiamo ritrovare una mutazione inattivante del gene GNAS, che anche in questo caso viene ereditata per via paterna.
La stessa sindrome di Albright può, come abbiamo già detto, essere dovuta allo PHPIa dove ritroviamo, oltre a tutti i rilievi, clinici e non, descritti a proposito dello PPHP, anche una specifica resistenza all’azione periferica di molti ormoni come il TSH, l’FSH, l’LH, l’ACTH e ritroviamo in circolo un aumentato livello di PTH proprio per la resistenza alla sua azione che poi in realtà è determinata dal mancato accoppiamento a cui è deputato il Gsα tra l’attività dell’adenilato ciclasi e il recettore di superficie per il PTH. Tornando a quanto detto precedentemente, è questa la forma che presenta trasmissione per via materna e abbiamo già notato come è proprio l’allele ereditato per via materna che è normalmente espresso in quasi tutti i tessuti ed è quindi proprio la sua mutazione quella che porta a maggiori danni.