

Il quadro Istopatologico
L’elemento che caratterizza l’istologia di questa patologia è l’ossificazione che si presenta di tipo diretto membranoso, senza una fase cartilaginea o elementi cartilaginei. Ritroviamo infatti osteociti, canali di Havers, “cement line”, osteoblasti e osteoclasti. L’ossificazione è presente estensivamente nel tessuto adiposo sottocutaneo, coinvolgendo fasce e muscoli sottostanti e soltanto focalmente coinvolge il derma. L’apparenza dell’ossificazione eterotopica è comunque abbastanza variabile. In alcune zone sembra essere composta da uno stroma cellulare altamente vascolarizzato in cui successivamente avviene l’ossificazione. Altre zone mostrano spicule ossee immerse in uno stroma fibroso con alcuni strati di ossificazione in periferia. Infine ci sono aree di osso perfettamente maturo, nel quale tutti gli elementi ben differenziati di midollo occupano lo spazio midollare.
Queste caratteristiche istopatologiche differenziano la POH dalla fibrodisplasia ossea progressiva in cui l’ossificazione è secondaria ad un processo infiammatorio ed è di tipo encondrale senza coinvolgimento del derma e del sottocute e con abbondante presenza di cartilagine.
Il quadro Radiologico
È molto caratteristico, in radiologia tradizionale le ossificazioni assumono un aspetto lacunare, a maglie di una rete, talora figurato a merletto, a bozzolo (cocoon degli autori anglosassoni) e questo corrisponde alle ramificazioni a tralci di vite che all’esame anatomico macroscopico vediamo infiltrarsi nei tessuti approfondendosi, seguendo nel loro decorso i fasci neuro – vascolo – connettivali.
Alla TAC possiamo notare un aspetto a bulbo di cipolla dato dall’interessamento delle fasce muscolari e dei setti connettivali nei gruppi muscolari (perimisio, endomisio).
La radiologia mostra ancora la normalità dello scheletro, l’assenza di malformazioni ossee primitive di qualsiasi genere a cominciare dalle anormalità falangee, e, eventualmente, la presenza di malformazioni secondarie alle ossificazioni, come scoliosi, incurvamento di ossa lunghe in arti la cui crescita è stata impedita dalla presenza delle ossificazioni, lussazioni e sublussazioni da pressione delle ossificazioni stesse, etc…
Una cosa che comunque viene sempre messa in evidenza è anche, soprattutto nelle fasi iniziali, il distacco delle ossificazioni patologiche dalle normali ossa dell’organismo.
Eziopatogenesi
L’eziologia e la patogenesi della POH rimangono sconosciute.
La POH è una malattia geneticamente determinata, la sua genetica è piuttosto complessa è probabilmente non completamente nota. Quella che per la maggioranza degli autori è implicata è una mutazione spontanea inattivante del gene GNAS, che è un gene che codifica per l’alfa subunità della proteina G – Gsα – che stimola l’adenilato ciclasi, con l’effetto di una diminuita attività della proteina stessa.
Questo gene è presente sul braccio lungo del cromosoma 20 al locus 20q13.2 – q13.3, la mutazione somatica inattivante avviene in fase post-zigotica e si esprime in un pattern a tipo mosaicismo, e si trasmette successivamente come autosomica dominante.
La proteina Gsα è implicata nella trasduzione di segnale a livello della membrana cellulare, ed è coinvolto nell’attività di molti ormoni. Le proteine G sono proteine leganti i nucleotidi guaninici ed interagiscono con il dominio citoplasmatico di recettori extracellulari per mediare molte vie di trasduzione di segnali; sono composte da 3 subunità: alfa, beta e gamma, quella che è interessata nel nostro caso è appunto la subunità alfa(Gsα).
Sebbene le vie ossee specifiche regolate dall’attivazione della proteina G rimangano da essere ancora chiarite, sono state identificate parecchie proteine G, accoppianti recettori agonisti, che influenzano la formazione ossea.
Nella sindrome di McCune – Albright (MIM 174800) mutazioni somatiche attivanti dello GNAS, che danno una sovrastimolazione dell’attività di Gsα, portano ad un arresto della maturazione degli osteoblasti e quindi a displasia fibrosa dell’osso scheletrico. Al contrario le mutazioni inattivanti dello GNAS nei pazienti con AHO, POC e POH portano ad una sottostimolazione delle vie metaboliche in cui è implicata l’attività di Gsα e ciò si concretizza in ossificazione eterotopica dei tessuti connettivi molli.
Questi dati, considerati insieme, suggeriscono che lo GNAS potrebbe normalmente funzionare come un inibitore della osteoblasto-sintesi.
La distribuzione a mosaico delle lesioni ha fatto pensare sin dal 1994 che nella patogenesi fosse implicata una mutazione delle cellule staminali mesenchimali.
Si è pensato che questa cellula o più cellule precursori siano presenti nella cute, tessuto adiposo sottocutaneo, muscoli, tendini e legamenti, e abbiano la potenzialità di dar luogo sia a cellule ossee che a diverse cellule mesenchimali tramite meccanismi di relais con sistemi di facilitazione/inibizione.
A questo proposito dobbiamo sottolineare che la formazione diretta membranosa di osso che avviene nel tessuto adiposo sottocutaneo in questi pazienti porta evidenze a favore di una stretta relazione tra adipogenesi e osteogenesi nei tessuti periferici, un fenomeno già ben documentato nelle cellule stromali del midollo osseo e nelle cellule progenitrici mesodermiche rese immortali.
Nel contesto su esposto il ruolo di Gsα è probabilmente quello di inibire la via ossea, una mutazione che intervenga alterando Gsα ne elimina l’inibizione sulla via ossea.
Bisogna comunque tener conto di alcune evidenze che ancora non hanno trovato una spiegazione univoca. In primis sono noti due casi in cui, pur essendo presente la mutazione del gene GNAS, non è presente alcuna manifestazione fenotipica, senza poi dire che questi soggetti hanno avuto figli ammalati (e quindi hanno trasmesso la mutazione!) e precisamente il primo ha avuto 3 figlie femmine affette da POH e 2 normali, il secondo 5 figlie affette ed una normale e in terza generazione da queste 5 figlie affette da POH si sono avuti 4 figli maschi affetti da sindrome di Albright (AHO), ma questo è un altro discorso che approfondiremo a breve. Invece, come spiegare la presenza di mutazione e assenza di patologia?
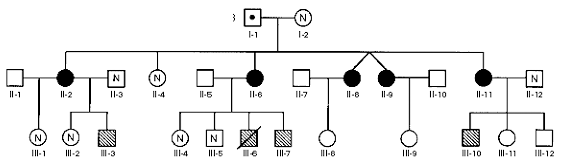
| Fig. 2: Albero genealogico: Simboli vuoti soggetti non indagati; simboli pieni affetti da POH; simboli tratteggiati affetti da AHO; N soggetti senza mutazione; punto centrale presenza di mutazione senza patologia. (da Shore et al.) |